国家药监局关于发布真实世界证据支持药物研发与审评的指导原则(试行)的通告(2020年第1号)
为进一步指导和规范真实世界证据用于支持药物研发和审评的有关工作,保障药物研发工作质量和效率,国家药品监督管理局组织制定了《真实世界证据支持药物研发与审评的指导原则(试行)》,现予发布。
特此通告。
真实世界证据支持药物研发与审评的
指导原则(试行)
一、引言
(一)背景与目的
随机对照试验(Randomized Controlled Trial,RCT)一般被认为是评价药物安全性和有效性的金标准,并为药物临床研究普遍采用。RCT严格控制试验入组、排除标准和其它条件,并进行随机化分组,因此能够最大限度地减少其它因素对疗效估计的影响,使得研究结论较为确定,所形成的证据可靠性较高。但RCT有其局限性:一是RCT的研究结论外推于临床实际应用时面临挑战,如严苛的入排标准使得试验人群不能充分代表目标人群,所采用的标准干预与临床实践不完全一致,有限的样本量和较短的随访时间导致对罕见不良事件探测不足等;二是对于某些疾病领域,传统RCT难以实施,如某些缺乏有效治疗措施的罕见病和危及生命的重大疾病;三是传统RCT或需高昂的时间成本。因此,在药物研发和监管领域如何利用真实世界证据(Real World Evidence,RWE)评价药物的有效性和安全性,已成为全球相关监管机构、制药工业界和学术界共同关注且具有挑战性的问题。
一是需要从概念上厘清真实世界证据的定义、范畴和内涵。
二是真实世界数据(Real World Data,RWD)是否适用于回答临床所关注的科学问题,所生成的真实世界证据能否或如何起到充分的支撑作用,涉及诸多亟待商榷和解决的问题,包括数据来源、数据标准、数据质量、数据共享、数据的基础建设等,也对指南的制定提出了迫切需求。
三是利用真实世界数据的方法学有待规范。真实世界证据源于对真实世界数据的正确和充分分析,所采用的分析方法主要是因果推断方法,涉及较复杂的模型、假设甚至人工智能和机器学习方法的应用等,对相关人员提出了更高的要求。
四是真实世界证据的适用范围有待明确。真实世界证据与传统RCT提供的证据均可以是药物监管决策证据的组成部分,支持监管决策形成综合、完整而严谨的证据链,从而提高药物研发和监管的科学性和效率。因此,需要根据药物研发和监管的现实情况明确真实世界证据的适用范围,并能够随现实情况变化进行调整。
鉴于上述情况,本指南旨在厘清药物研发和监管决策中真实世界证据的相关定义,指导真实世界数据收集以及适用性评估,明确真实世界证据在药物监管决策中的地位和适用范围,探究真实世界证据的评价原则,为工业界和监管部门利用真实世界证据支持药物监管决策提供参考意见。本指导原则仅代表当前的观点和认识,随着研究和认识的深入将不断修订和完善。
(二)国内外监管机构在法规或指南制定方面的进展
2009年美国复苏与再投资法案对实效比较研究(Comparative Effectiveness Research,CER)起到了巨大推动作用。基于CER的真实世界环境的背景,真实世界研究(Real World Research/Study,RWR/RWS)得以更广泛的应用。
美国于2016年12月通过《21世纪治愈法案》,鼓励美国食品药品监督管理局(The Food and Drug Administration,FDA)开展研究并使用真实世界证据支持药物和其它医疗产品的监管决策,加快医药产品开发。在该法案的推动下,2017-2019年FDA先后发布了《使用真实世界证据支持医疗器械监管决策》《临床研究中使用电子健康档案数据指南》《真实世界证据计划的框架》和《使用真实世界数据和真实世界证据向FDA递交药物和生物制品资料》。
欧盟药品管理局(European Medicines Agency,EMA)于2013年参与的GetReal Initiative项目,致力于开发出收集与综合RWE的新方法,以便更早地用于药品研发和医疗保健决策过程中。EMA于2014年启动了适应性许可试点项目,探索利用真实世界数据包括观察性研究数据等用于监管决策的可行性。2017年药品局总部(Heads of Medicines Agencies,HMA)与EMA联合成立大数据工作组,旨在使用大数据改进监管决策并提高证据标准,其中RWE是大数据的一个子集,包括电子健康档案、登记系统、医院记录和健康保险等数据。
日本药品和医疗器械管理局(PMDA)在国际人用药品注册技术要求协调会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)层面提出更高效利用真实世界数据开展上市后药物流行病学研究的技术要求新议题。
事实上,全球使用真实世界数据对医疗产品进行安全性评价已经积累了丰富的实践经验,例如2008年美国FDA启动了哨点计划,利用现有的电子医疗健康数据实现对上市后医疗产品安全性的主动监测。
我国系统性开展使用真实世界证据支持药物监管决策的工作尚处于起步阶段。国家药品监管部门在审评审批实践中开始应用真实世界证据,相关示例参见附2。
二、真实世界研究的相关定义
真实世界研究是指针对预设的临床问题,在真实世界环境下收集与研究对象健康有关的数据(真实世界数据)或基于这些数据衍生的汇总数据,通过分析,获得药物的使用情况及潜在获益-风险的临床证据(真实世界证据)的研究过程(如图1所示)。
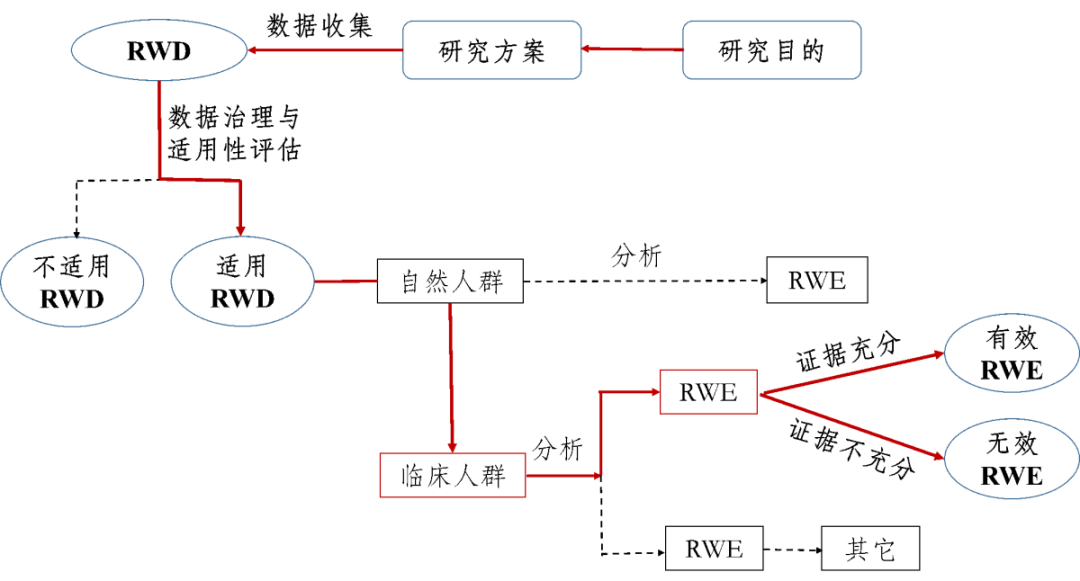
图1支持药物监管决策的真实世界研究路径(实线所示)
真实世界研究所产生的真实世界证据既可用于支持药物研发与监管决策,也可用于其它科学目的(如不以注册为目的的临床决策等)。本指南主要用于支持药物监管决策、以临床人群为研究对象的真实世界研究,个别情形下也会涉及更广泛的自然人群,如疫苗等健康人群的预防用药。
真实世界研究的类型大致分为非干预性(观察性)研究和干预性研究。前者包括不施予任何干预措施的回顾性和前瞻性观察性研究,患者的诊疗、疾病的管理、信息的收集等完全依赖于日常医疗实践;后者与前者最大的不同是主动施予某些干预措施,如实用临床试验(Pragmatic Clinical Trial,PCT)等。由于真实世界研究的多样性、设计的复杂性、分析方法的高要求和对结果解释的不确定性,对药物的安全性和有效性的评价以及监管决策提出了更高的要求。
(一)真实世界数据
1.定义
真实世界数据是指来源于日常所收集的各种与患者健康状况和/或诊疗及保健有关的数据。并非所有的真实世界数据经分析后都能成为真实世界证据,只有满足适用性的真实世界数据才有可能产生真实世界证据。
2.真实世界数据的来源
真实世界数据的常见来源包括但不限于:
(1)卫生信息系统(Hospital Information System,HIS):类似于电子健康档案,包括结构化和非结构化的患者记录,如患者的人口学特征、临床特征、诊断、治疗、实验室检查、安全性和临床结局等。
(2)医保系统:包含患者基本信息、医疗服务利用、诊断、处方、结算、医疗付费和计划保健等结构化字段的数据。
(3)疾病登记系统:特定疾病(通常是慢性病)患者的数据库,通常来源于医院的疾病人群队列登记。
(4)国家药品不良反应监测哨点联盟(China ADR Sentinel Surveillance Alliance,CASSA):利用医疗机构电子数据建立药品及医疗器械安全性的主动监测与评价系统。
(5)自然人群队列和专病队列数据库:国内已经建立或正在建立的自然人群队列和专病队列数据库。
(6)组学相关数据库:采集患者的生理学、生物学、健康、行为和可能的环境相互作用的组学相关信息,如药物基因组学、代谢组学和蛋白质组学的数据库。
(7)死亡登记数据库:由医院、疾病预防控制中心和户籍部门联合确认的死亡登记所形成的数据库。
(8)患者报告结局数据:由患者自行填报的自我评估或测量的数据。
(9)来自移动设备端的数据:应用医用移动设备,如可穿戴设备,检测受试者获得的相关数据。
(10)其他特殊数据源:部分地区医疗机构根据相关政策、法规,因临床急需进口少量境外已上市药品等用于特定医疗目的而生成的有关数据;为特殊目的创建的数据库,如法定报告传染病数据库、国家免疫规划数据库等。
3.数据标准
统一的数据标准使递交的资料具有可预测性和一致性,并能与其它数据库之间共享信息。递交的数据应当在数据标准的规划、数据的采集和编码及储存、分析数据的格式、数据的核查和可溯源性、电子递交的格式等方面有统一的标准。
(二)数据的适用性
真实世界数据的适用性主要通过数据相关性和可靠性进行评估。
1.相关性
评估真实世界数据是否与所关注的临床问题密切相关,其重要因素包括但不限于:
(1)是否包含与临床结局相关的重要变量和信息,如药物暴露、患者人口学和临床特征、协变量、随访时间、结局变量等;
(2)临床结局定义是否准确,相应的临床意义是否明确;
(3)真实世界数据中的患者对于研究的目标人群是否具有代表性;
(4)是否有足够的样本量以及随访时间以证明疗效并获取充分的潜在安全性事件。
2.可靠性
真实世界数据的可靠性主要从数据的完整性、准确性、透明性和质量保证方面进行评价。
(1)完整性:真实世界数据无法避免数据缺失问题,包括变量的缺失和变量值的缺失。当数据缺失比例超过一定限度时,尤其涉及研究的关键变量时,例如影响研究结局的诸多重要预后协变量缺失或变量值缺失,会加大研究结论的不确定性,此时,需要慎重考虑该数据能否支持产生真实世界证据。
(2)准确性:数据的准确性极为重要,通常需要参照较权威的数据来源进行识别或验证。数据元素和转化数据的算法均应保证其正确。数据的准确性还反映在数据的一致性和合理性上,一致性包括数据库内部的相关数据标准、格式和计算方法等必须一致;合理性包括变量数值的唯一性、合理的区间和分布、相关变量的预期依从关系以及时变型变量是否按预期改变等。
(3)透明性:数据的来源、收集与治理的全过程应透明、清晰,并具有可溯源性,尤其是关键的暴露、协变量以及结局变量等应能追溯到源数据。数据的透明性还包括数据的可及性、数据库之间的信息共享和对患者隐私的保护方法的透明。
(4)质量保证:真实世界数据的可靠性需考虑数据质量,质量保证的措施包括但不限于:数据收集是否有明确流程和合格人员;是否使用了共同定义框架,即数据字典;是否遵守采集关键数据点的共同时间框架;是否建立与收集真实世界数据有关的研究计划、协议和分析计划的时间安排;用于数据元素采集的技术方法是否充分,包括各种来源数据的集成、药物使用和实验室检查数据的记录、随访记录、与保险数据的链接以及数据安全等。
(三)真实世界证据
真实世界证据是指通过对适用的真实世界数据进行恰当和充分的分析所获得的关于药物的使用情况和潜在获益-风险的临床证据,包括通过对回顾性或前瞻性观察性研究或者实用临床试验等干预性研究获得的证据。
三、真实世界证据支持药物监管决策
真实世界证据应用于支持药物监管决策,涵盖上市前临床研发以及上市后再评价等多个环节。例如,为新产品批准上市提供有效性或安全性的证据;为已获批产品修改说明书提供证据,包括增加或修改适应症,改变剂量、给药方案或给药途径,增加新适用人群,增加实效比较信息,增加安全性信息等;作为上市后要求的一部分支持监管决策的证据等。
下面是真实世界证据支持药物监管决策的某些应用范围,但并不排除其它合理的应用。
(一)为新药注册上市提供有效性和安全性的证据
根据不同疾病的特征、治疗手段的可及性、目标人群、治疗效果和其它与临床研究相关的因素等,可以通过真实世界研究获得药物的效果和安全性信息,为新药注册上市提供支持性证据。
常见的为新药注册上市提供有效性和安全性证据的真实世界研究有:使用真实世界数据获得的结局或安全性数据的随机临床试验,包括PCT设计等;以及针对某些缺乏有效治疗措施的罕见病和危及生命的重大疾病,而采用基于真实世界证据作为外部对照的单臂临床试验。
(二)为已上市药物的说明书变更提供证据
对于已经上市的药物,新增适应症通常情况下需要RCT支持。但当RCT不可行或非最优的研究设计时,采用PCT或观察性研究等生成的真实世界证据支持新增适应症可能更具可行性和合理性。
在儿童用药等领域,利用真实世界证据支持适应症人群的扩大也是药物监管决策可能适用的情形之一。
总的来说,真实世界证据支持已上市药物的说明书变更主要包括以下几种情形:
1.增加或者修改适应症;
2.改变剂量、给药方案或者用药途径;
3.增加新的适用人群;
4.添加实效比较研究的结果;
5.增加安全性信息;
6.说明书的其它修改。
(三)为药物上市后要求或再评价提供证据
基于RCT证据获批的药物,通常由于病例数较少、研究时间较短、试验对象入组条件严格、干预标准化等原因,存在安全性信息有限、疗效结论外推不确定、用药方案未必最优、经济学效益缺乏等不足,需要利用真实世界数据对药物在真实医疗实践中的效果、安全性、使用情况,以及经济学效益等方面进行更全面的评估,并不断根据真实世界证据做出决策调整。
(四)名老中医经验方、中药医疗机构制剂的人用经验总结与临床研发
对于名老中医经验方、中药医疗机构制剂等已有人用经验药物的临床研发,在处方固定、生产工艺路线基本成型的基础上,可尝试将真实世界研究与随机临床试验相结合,探索临床研发的新路径。
应用真实世界证据支持已有人用经验中药的临床研发策略可以有多种,应根据产品的特点、临床应用情况以及数据适用性等方面的考虑,选择不同的研发策略。例如可以探索将观察性研究(包括回顾性和前瞻性)代替常规临床研发中I期和/或II期临床试验,用于初步探索临床疗效和安全性;在观察性研究的基础上,再通过RCT或PCT进一步确证已有人用经验中药的有效性,为产品的注册上市提供支持证据。如果经过评价,存在适用的高质量真实世界数据,且通过设计良好的观察性研究形成的真实世界证据科学充分,也可与药品监管部门沟通,申请直接作为支持产品上市的依据。
针对观察性研究与RCT或PCT研究相结合的研发策略,其实现也可以有多种路径,图2和图3是可能路径中的两种,但不限于此。图2是观察性研究与RCT研究相结合的路径,第一阶段先开展回顾性观察性研究,此阶段应尽可能地收集既往与使用该药品有关的真实世界数据,包括所有可能的协变量;制定数据清理规则;选择可能的对照;对数据质量进行评估;采用恰当的统计方法进行全面详细的分析。如果通过回顾性观察性研究得出该药品在临床应用中对患者具有潜在获益,可以进入下一研究阶段,否则研究终止。第二阶段开展前瞻性观察性研究。由于有了第一阶段的研究基础,该阶段可以将前瞻性观察性研究设计得更加周密,包括数据的采集及其系统、数据的质量控制、数据清理的规则、明确定义对照等。在前瞻性观察性研究进展到某一时期,如果数据分析结果与回顾性观察性研究结果一致,且继续显现出该药品在临床应用中对患者具有明显获益,可适时平行开展第三阶段的RCT研究。RCT研究可以先进行探索性RCT研究,但如果前期的观察性研究证据较充分,也可以直接进行确证性RCT研究。从时间上看,RCT研究的周期可被前瞻性观察性研究所覆盖,后者可以在RCT研究开始前结束,也可与RCT研究同时结束,甚至在RCT研究结束后继续延展一段时间,以积累更充分的真实世界证据,或用于其他目的,如增加适应症或扩大适用人群范围等。
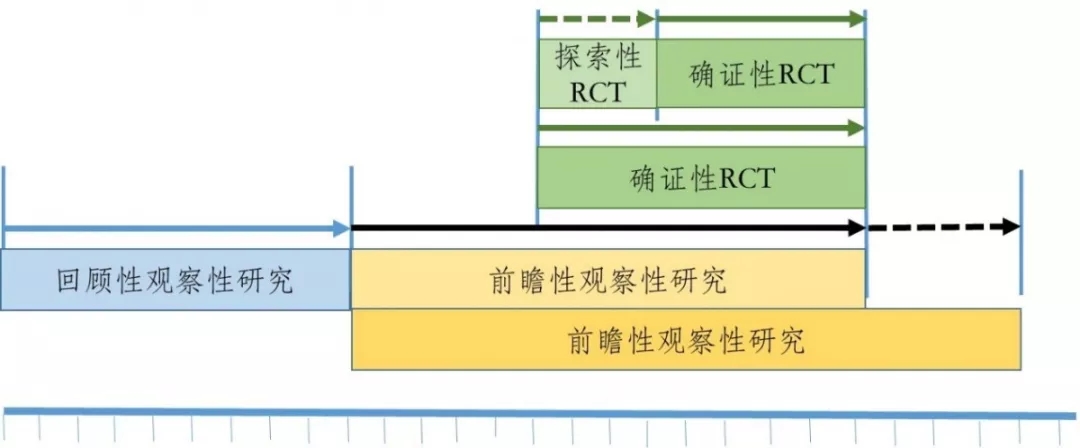
图2 已有人用经验中药临床研发的路径之一
观察性研究与PCT研究相结合的路径如图3所示,第一阶段先开展回顾性观察性研究,如果得出该药品在临床应用中对患者具有潜在获益,可以进入下一研究阶段,否则研究终止。第二阶段开展PCT研究,它所提供的证据可以用于支持其临床有效性和安全性的评价。
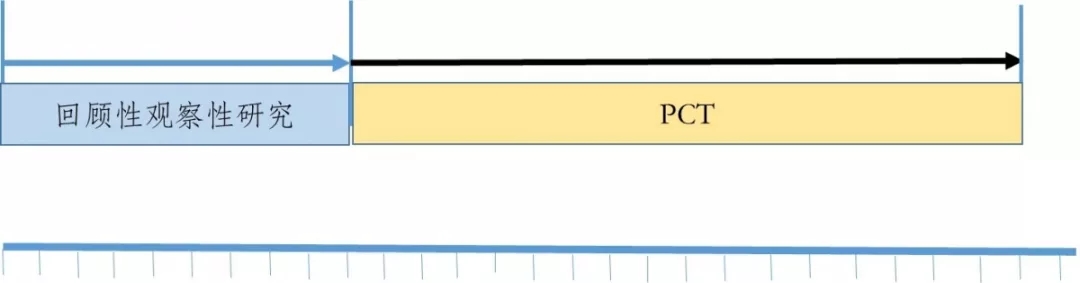
图3 已有人用经验中药临床研发的路径之二
已有人用经验中药的临床研发应根据产品的特点、基础研究的信息(如毒理试验)、临床应用情况、既往临床实践的有效数据积累等采取恰当的策略,并不局限于上述两种可能的策略。
(五)真实世界证据用于监管决策的其它应用
1.指导临床研究设计
利用真实世界证据指导临床研究设计有着现实的用途。例如,前述两种中药临床研发的路径,都采用了回顾性观察性研究所产生的真实世界证据,包括疾病的自然史、疾病在目标人群的流行率、标准化治疗的疗效和效果以及与疗效和效果有关的关键协变量在目标人群中的分布和变化等,为下一阶段的研究设计提供了依据。更为普遍的应用是真实世界证据可为入选和排除标准、样本量估计的参数、非劣效界值的确定等提供有效的参考依据,有助于审评中对设计合理性的判断。
2.精准定位目标人群
精准医疗旨在更好地预测药物对特定人群(亚组)的治疗获益和风险,基于真实世界数据的真实世界证据为精准医疗提供了可能。例如,传统临床试验因样本量有限,往往在研究计划中忽略或无暇顾及亚组效应,使得潜在的治疗应答者或具有严重副作用的高风险人群的重要信息不能充分体现,从而导致目标人群失准。由于真实世界数据往往是不同类型的大数据,通过详尽的分析,可以充分考察不同亚组的治疗获益和风险,进而得到真实世界证据以支持更精准的目标人群定位。
对于靶向治疗药物的临床前和早期临床研究,生物标记物的识别甚为关键。利用人群队列中的组学数据、公共基因库信息以及相关的临床资料等真实世界数据,通过多种机器学习类的目标靶向分析技术得到真实世界证据,可以支持靶向治疗药物的精确人群定位。
四、真实世界研究的基本设计
(一)实用临床试验
实用临床试验又称实操临床试验和实效临床试验,是指尽可能接近真实世界临床实践的临床试验,是介于RCT和观察性研究之间的一种研究类型。与RCT不同的是:PCT的干预既可以是标准化的,也可以是非标准化的;既可以采用随机分组方式,也可以自然选择入组;受试病例的入选标准较宽泛,对目标人群更具代表性;对干预结局的评价不局限于临床有效性和安全性;PCT一般使用临床终点,而避免使用传统RCT中可能使用的替代终点;可以同时考虑多个对照组,以反映临床实践中不同的标准化治疗;一般不设安慰剂对照;在大多数情况下不采用盲法,但对于如何估计和纠正由此产生的测量偏倚,需给予足够的重视;数据的收集通常依赖于患者日常诊疗记录。与观察性研究不同的是,PCT是干预性研究,尽管其干预的设计具有相当的灵活性。
例如,一项以患者为中心的、评价不同剂量阿司匹林的获益和长期有效性的研究采用了随机化的PCT设计,研究纳入患有动脉粥样硬化性心血管疾病且具有高风险缺血事件的患者,随机分配到两个不同剂量的阿司匹林治疗组(外加日常医疗保健),主要终点为来自电子健康档案和保险索赔数据库的全因死亡、非致死性心梗导致的住院以及由中风引起的住院的复合终点。
设计PCT时还应考虑以下因素:①收集到的数据是否适用于支持产生真实世界证据;②治疗领域和干预措施等是否符合各种形式的常规临床实践;③是否具有足够的可以用于评价的病例数(特别是临床结局罕见的情况);④参与PCT的各试验中心甚至不同的数据库之间对终点的评价和报告方法是否一致;⑤是否采用随机化方法控制偏倚;⑥当盲法不可行时,应考虑非盲对结局变量(特别是患者报告的结局)可能产生的影响,可使用不受治疗分组影响的终点(如中风、肿瘤大小等),以减少非盲带来的可能偏倚。
由于PCT需要考虑所有可能的潜在因素的影响,包括各种偏倚和混杂因素的影响,故其研究设计和统计分析较为复杂,所需的样本量通常远超RCT设计。PCT如果采用随机化方法将减小混杂因素的影响从而提供稳健的因果推断。由于是在更接近真实临床实践环境下开展的研究,PCT所获得的证据在多数情况下被视为是较好的真实世界证据。
(二)使用真实世界证据作为外部对照的单臂试验
单臂临床试验也是验证研究药物有效性和安全性的一种方法。例如,针对某些罕见病的临床试验,由于病例稀少导致招募困难;针对某些缺乏有效治疗措施的危及生命的重大疾病,随机对照试验往往存在伦理问题。因此,以上两种情况可以考虑以自然疾病队列形成的真实世界数据作为外部对照的基础。
外部对照主要用于单臂试验,可以是历史对照也可以是平行对照。历史外部对照以早先获得的真实世界数据作为对照,需考虑不同历史时期对疾病的定义、诊断、分类、自然史和可用的治疗手段等对可比性的影响;平行外部对照则是将与单臂试验同期开展的疾病登记数据作为对照。采用外部对照需考虑目标人群的可比性对真实世界证据的影响;对于接受其它干预措施的病人的数据,应考虑是否有足够的协变量以支持正确和充分的统计分析。
使用外部对照具有局限性,主要包括医疗环境不同、医疗技术随时间变化、诊断标准不同、结局的测量和分类不同、患者的基线水平不同、干预多样化、数据质量难以保证等。这些局限使得研究对象的可比性、研究结果的精确性、研究结论的可靠性和外推性等均面临挑战。
为克服或减少这些局限,一是要确保所采集的数据符合真实世界数据的适用性要求。二是采用平行外部对照设计要优于历史对照,平行外部对照可采用疾病登记模式,保障数据记录尽可能完整、准确。三是采用恰当的统计分析方法,如合理利用倾向评分(Propensity Scores,PS)方法,虚拟匹配对照方法等。四是要充分使用敏感性分析和偏倚的定量分析来评价已知或已测的混杂因素和未知或不可测量的混杂因素以及模型假设对分析结果的影响。
(三)观察性研究
观察性研究所采集的数据接近真实世界,其最主要的局限在于存在各种偏倚、数据质量难以保证、已知或已测和未知或不可测量的混杂因素较难识别等,使得研究结论具有很大的不确定性。
观察性研究所收集的数据是否适合产生真实世界证据,以支持监管决策,关注要点至少应包括:①数据特征:例如,数据来源及其质量、研究的人群、暴露和相关终点的数据采集、记录的一致性、数据治理过程、缺失数据的描述等;②研究设计和分析:例如,有无合适的阳性对照,是否考虑了潜在未测或不可测混杂因素以及可能的测量结果的变异,分析方法是否严谨、透明且符合监管要求等;③结果的稳健性:为保证结果的稳健性,预先确定了何种敏感性分析、偏倚定量分析和统计诊断方法。
观察性研究的主要分析方法是因果推断(见附3)。
五、真实世界证据的评价
评价真实世界证据应依从两个主要原则:真实世界证据是否可以支持需要回答的临床问题;已有的真实世界数据是否可以通过科学的研究设计、严谨的组织实施及合理的统计分析得到所需的真实世界证据。
(一)真实世界证据和其所支持的临床问题
在决定使用包括真实世界证据在内的任何证据之前,首先应明确需要回答的临床问题。例如,药品上市后和其它药品联合使用的安全性考虑;已获批产品的新增适应症研究;为某罕见病的单臂临床试验建立稳健可靠的历史或者外部对照等。其次需要考虑使用真实世界证据是否能够回答面对的临床问题,应从科学方面的有效性(例如,科学上的可解释性、假设的合理性、I类误差控制等)、监管要求(是否与其他监管要求冲突、有无特殊疾病领域的监管要求等)、伦理方面的问题(如果不使用真实世界证据是否会带来伦理问题)和可操作性(例如,是否有独立统计师以及确保统计师对结局变量的盲态,以避免匹配时可能带来的偏倚;是否有其他操作上的挑战等)四个方面评价。以上问题综合考虑,是衡量真实世界证据应用的重要准则。
(二)如何从真实世界数据到真实世界证据
一般至少应考虑以下几点:①研究环境和数据采集接近真实世界,如更有代表性的目标人群,符合临床实践的干预多样化,干预的自然选择等;②合适的对照;③更全面的效果评价;④有效的偏倚控制,如随机化的使用,测量和评价方法的统一等;⑤恰当的统计分析,如因果推断方法的正确使用、合理的缺失数据处理、充分的敏感性分析等;⑥证据的透明度和再现性;⑦合理的结果解释;⑧各相关方达成共识。
需要特别注意的是,所有与产生真实世界证据相关的研究设计、假设以及具体定义,均应事先在研究方案中明确阐述。事后补充的数据引用、定义、分析以及解释,通常不能用于监管决策。
六、与审评机构的沟通交流
以药品注册为目的使用真实世界证据,需要与药品审评部门进行充分的沟通交流,以确保双方对使用真实世界证据以及开展真实世界研究等方面达成共识。
申请人计划使用真实世界证据支持药品注册事项时,在研究实施前,应当按照药品审评部门的沟通交流途径主动提出沟通交流申请,就研究目标、真实世界证据使用的可行性、研究设计、数据收集和分析方法等方面进行书面或会议的沟通与讨论。
申请人完成真实世界研究后,计划递交申报资料前,也应当申请与审评部门进行沟通交流,就研究的实施情况、研究结果与结论、申报资料要求等内容进行沟通确认。
【参考文献】
1. 孙宇昕,魏芬芳,杨悦. 真实世界证据用于药械监管与卫生决策的机遇与挑战. 中国药物警戒,2017,14(6):353-358.
2. 吴一龙,陈晓媛,杨志敏等(吴阶平医学基金会,中国胸部肿瘤研究协作组). 真实世界研究指南. 2018.
3. 中共中央办公厅,国务院办公厅. 关于深化审评审批制度改革鼓励药品医疗器械创新的意见. 2017.
4. ADAPTABLE Investigators. Aspirin Dosing: a Patient-Centric Trial Assessing Benefits and Long-Term Effectiveness (ADAPTABLE)study protocol. http://pcornet. org/wp-content/uploads/2015 /06/ADAPTABLE-Protocol-Final-Draft-6-4-15_for-post_06-26-. pdf[J]. Published June,2015,5.
5. Berger M,Daniel G,Frank K,et al. A frame work for regulatory use of real-world evidence[J]. White paper prepared by the Duke Margolis Center for Health Policy,2017,6.
6. Cave A,Kurz X,Arlett P. Real-world data for regulatory decision making: challenges and possible solutions for europe[J]. Clinical pharmacology and therapeutics,2019,106(1): 36.
7. Dreyer NA. Advancing a framework for regulatory use of real-world evidence: when real is reliable[J]. Therapeutic innovation & regulatory science,2018,52(3): 362-368.
8. Egger M,Moons K G M,Fletcher C,et al. GetReal: from efficacy in clinical trials to relative effectiveness in the real world[J]. Research synthesis methods,2016,7(3): 278-281.
9. Ford I,Norrie J. Pragmatic trials[J]. N Engl J Med,2016,375(5): 454-463.
10. Institute of Medicine 2009. Initial national priorities for comparative effectiveness research. Washington,DC: The National Academies Press. https://doi.org/10.17226/12648.
11. James S. Importance of post-approval real-word evidence[J]. European Heart Journal-Cardiovascular Pharmacotherapy,2018; 4(1):10-11.
12. Kohl S. Joint HMA/EMA task force on big data established[J]. Eur J Hosp Pharm,2017,24(3): 180-190.
13. Lash TL,Fox MP,Fink AK. Applying quantitative bias analysis to epidemiologic data[M]. Springer Science & Business Media,2011.
14. Makady A,de Boer A,Hillege H,et al. What is real-world data? A review of definitions based on literature and stakeholder interviews[J]. Value in health,2017,20(7): 858-865.
15. Olariu E,Papageorgakopoulou C,Bovens S M,et al. Real world evidence in Europe: a snapshot of its current status[J]. Value in Health,2016,19(7): A498.
16. Roland M,Torgerson D J. Understanding controlled trials: What are pragmatic trials?[J]. BMJ,1998,316(7127): 285.
17. Sherman RE,Anderson SA,Dal Pan GJ,et al. Real-world evidence—what is it and what can it tell us[J]. N Engl J Med,2016,375(23): 2293-2297.
18. Sugarman J,Califf RM. Ethics and regulatory complexities for pragmatic clinical trials[J]. JAMA,2014,311(23): 2381-2382.
19. US Food and Drug Administration. Framework for FDA’s real-world evidence program. December 2018[J]. 2019.
20. Velentgas P,Dreyer NA,Nourjah P,Smith SR,Torchia MM,eds. Developing a Protocol for Observational Comparative Effectiveness Research: A User’s Guide. AHRQ Publication No. 12(13)-EHC099. Rockville,MD: Agency for Healthcare Research and Quality; January 2013. www.effectivehealthcare.ahrq.gov/ Methods-OCER.cfm.
21. Von Elm E,Altman D G,Egger M,et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE)statement: guidelines for reporting observational studies[J]. Annals of internal medicine,2007,147(8): 573-577.