药物相互作用研究指导原则
一、引言
本指导原则旨为拟进行药物(指新药,包括生物制品)相互作用研究的申办方提供建议。本指导原则反映了国家食品药品监督管理局(以下简称SFDA)审评机构的当前认识:即新药的代谢应该在药物研发过程中进行确定,该药与其他药物之间的相互作用应作为安全性和有效性评价的一部分进行研究。本指导原则建议的研究方法是基于以下的共识,即:是否应进行某项特定的试验取决于药物的特征及拟定的适应证。药物相互作用除了发生在代谢过程中外,也可能发生在吸收、分布和排泄过程。目前,越来越多的报告显示药物相互作用与转运体相关,因此,它们也是新药开发过程中应该考察的因素之一。药物相互作用还可能改变药代动力学/药效动力学(PK/PD)的相互关系。
二、背景
( 一 )代谢
药物在作用部位的浓度所引起预期的和非预期的效应通常与用药剂量或血药浓度有关,而血药浓度受到药物吸收、分布、代谢/或排泄的影响。药物或其代谢产物的消除通常通过两种途径:即代谢(常在肝脏或肠粘膜)和排泄(常在肾和肝脏)。此外,治疗用蛋白制剂可通过与细胞表面受体产生特异性结合,然后经由细胞内吞和细胞内的溶酶体降解进行消除。肝脏消除主要由位于肝细胞内质网的细胞色素P450酶系,但也可经由非P450酶系系统,如通过N-乙酰基和葡萄糖醛酰转移酶完成。许多因素可影响药物在肝脏和肠内的代谢,如疾病、合并用药(包括中草药)、甚至食物(如西柚汁)等。虽然这些因素中的大多数通常可保持相对的稳定,但是合并用药往往会突然改变药物的代谢,因此需要特别关注。如果药物(包括前体药物)代谢成一种或多种活性代谢物,合并用药对药物代谢的影响就变得更为复杂。这种情况下,药物/药物前体的安全性和有效性不仅仅取决于原形药物的暴露量,还同时取决于其活性代谢物的暴露量,而活性代谢物的暴露量与其生成、分布和消除相关。因此,对新药安全性和有效性的评价应该包括药物的代谢情况以及该代谢对整个消除过程的贡献大小。基于此,在药物代谢和相互作用研究中,建立灵敏的、专属性强的药物及其重要代谢产物的测定方法具有重要的意义。
(二)药物相互作用
1.代谢相关的药物相互作用
许多药物的代谢消除(包括大部分通过P450酶系的代谢),可因合并用药而受到抑制、激活或诱导。因药物相互作用引起代谢的变化会相当大,可能导致药物或其代谢物在血液或组织中浓度水平以一个数量级或以上的降低或升高,也可能导致毒性代谢物的生成或毒性原型药物暴露量水平的升高。这些暴露量水平的较大变化可使一些药物和/或其活性代谢物的安全性和有效性特征发生重要的变化。此种变化不仅对于窄治疗窗(NTR)的药物最为明显,也最容易预期,而且对于非窄治疗窗(non-NTR)药物有时也可能发生(例如HMG CoA还原酶抑制剂)。
代谢相关的药物相互作用研究的重要目的是探索新药是否有可能对已上市的、并可能在医疗诊治中合用的药物的代谢消除产生显著影响。此外,也应当探索已上市药物是否可能对新药的代谢消除产生影响。本身并不被广泛代谢的药物也可能对合用药物的代谢产生重要作用,因此,即使对于代谢不是主要消除途径的新药,也应进行代谢相关的药物相互作用的探索。
尽管某些治疗用生物制品会改变经P450酶代谢的药物代谢过程(例如,I 型干扰素在转录和转录后水平抑制CYP1A2的生成,从而可抑制茶碱的清除),然而,典型的生物转化研究不是治疗性生物制品评价的普遍要求。随着治疗用蛋白制剂临床使用的增加,可能会引起其对药物代谢潜在影响的担忧。通常无法通过体外试验发现此类相互作用。鉴于其独特的性质,开始进行生物制品有关的药物相互作用研究之前,建议适当与SFDA进行商讨。
根据遗传多态性或其他易于鉴别的因素(如年龄、种族和性别),识别出不同患者人群中的药物代谢差异,会有助于对代谢相关的药物相互作用研究结果的诠释。这些因素(如CYP2D6基因型)可能影响相互作用的程度。此外,对于缺乏主要清除途径的受试者,其他的代谢途径就变得非常重要,应进行研究。
代谢相关的药物相互作用研究的一个特殊目标是确定这种相互作用是否足以导致必须对该药自身或与其合用药物的剂量进行调整、或者合用时需要进行额外的治疗监测。
在一些例子中,存在药物相互作用的情况下,了解如何调整剂量或给药方案、或如何避免发生相互作用,对即使存在药物相互作用且会产生不可接受毒性的药物也会给予上市许可。有时,可有目的地利用药物相互作用来提高某一种药物的暴露水平或减少其消除(如利托纳韦和络匹那韦)。少见的情况下,某种药物对其他药物产生的相互作用或其他药物对其代谢的影响造成该药不能安全上市应用。
2.转运体相关的药物相互作用
与转运体相关的药物相互作用的文献越来越多,其中的实例包括转运体的抑制或诱导,如P-糖蛋白(P-gp)、有机阴离子转运体(OAT)、有机阴离子转运多肽(OATP)、有机阳离子转运体(OCT)、多药耐药相关蛋白(MRP)、和乳腺癌耐药蛋白(BCRP)。有关与转运体的相互作用实例包括地高辛与奎尼丁、非索非那定与酮康唑(或红霉素)、青霉素与丙磺舒、以及多非利特与西咪替丁等。在各种转运体中,P-gp是研究最充分的转运体,可在新药开发中用于评价药物相互作用。
三、一般策略
药物开发应尽可能遵循这样一种顺序,即早期的体外和体内研究可完整阐述某个受关注的问题或者提供信息指导进一步的研究。较好的情况是对研究依次规划,从体外研究到人体的体内研究。如有必要,可根据情况采用特殊的研究设计和方法学。在许多情况下,从早期体外和早期临床试验中获得的阴性观察结果,可免除进行后期的临床相互作用研究的必要性。早期研究应探索药物主要通过排泄还是代谢进行消除,对于后一种情况应确定主要的代谢途径。在开发的早期阶段可采用适当的体外探针,仔细筛选可能发生相互作用的药物,用于早期的体内相互作用研究。可在开发的早期阶段进行药物相互作用研究,必要时可在后期的开发阶段进一步评估已经观察到的相互作用。早期临床研究还可获得普通人群、特殊人群和个体的剂量、血药浓度和效应的相互关系信息,这些信息有助于对代谢相关的药物相互作用研究的结果进行阐释。如果根据体外/体内研究发现潜在的药物相互作用,在具备可行性的情况下,鼓励申办者设计较大的临床试验并对获得的安全性和有效性数据库进行调查分析,以便于:(1)确认或发现早期研究探测到的相互作用,和/或(2)验证针对潜在相互作用进行的剂量调整或用药方法的其他改变是否可有效地避免药物相互作用的不良后果。
(一)体外研究
体外研究与体内代谢相关的药物相互作用研究结果之间的定量关系,目前还不完全清楚。但是可以将体外研究作为筛选方法,以获得代谢途径信息和排除某种重要的代谢途径及通过该途径发生药物相互作用,避免进行不必要的体内研究。这种可能应当建立在采用经过妥善验证的试验方法以及合理选择底物/发生相互作用药物浓度的基础上。
相反,如在体外的代谢相关药物相互作用研究中获得阳性结果,由于体外发现尚不足以对某种代谢途径或相互作用的临床重要性进行准确地定量估计,因此建议进行临床试验。虽然体外研究可揭示抑制作用的出现与否,但识别诱导发生的能力有限。鉴于这一原因,关于合用药物诱导代谢途径的信息,体内试验一直是其主要的手段。
例如,在治疗浓度下,体外研究显示CYP1A2、CYP2C8、CYP2C9、CYP2C19、CYP2D6、或CYP3A酶系不参与研究药物的代谢,则将无需进行评价CYP2D6抑制剂或CYP1A2、CYP2C8、CYP2C9、CYP2C19、或CYP3A抑制剂/诱导剂影响此药物消除的临床研究。
同样,如果体外研究结果表明所研究的药物对CYP1A2、CYP2C8、CYP2C9、CYP2C19、CYP2D6、或CYP3A代谢无抑制作用,那么就不需要进行相应体内的研究,即无需在这些酶抑制水平下进行研究药物与经这些酶消除的合用药物的体内相互作用研究。
CYP2D6酶未显示有可诱导性。最新数据显示CYP2C、CYP2B和ABCB1(P-gp)转运体与CYP3A具有协同诱导作用,CYP3A似乎对所有已知协同诱导物均敏感。因此,为了评价研究药物是否对CYP1A2、CYP2C8、CYP2C9、CYP2C19、或CYP3A有诱导作用,最初体外诱导评价可能仅包括CYP1A2和CYP3A。如果体外研究结果表明研究药物对CYP3A代谢不具有诱导作用,那么就不需要在这些酶诱导水平下进行研究药物与经CYP2C/CYP2B及CYP3A消除的合用药物的体内相互作用研究。
CYP2B6介导的药物相互作用是重要的相互作用。适当时,需进行基于该酶的相互作用的体外研究。其他CYP酶(包括CYP2A6和CYP2E1)被认为较少参与具有临床重要性的药物相互作用,只有在必要时,才考虑进行相关研究。
图 1 提供了与CYP酶有关的药物相互作用研究的决策树作为参考。
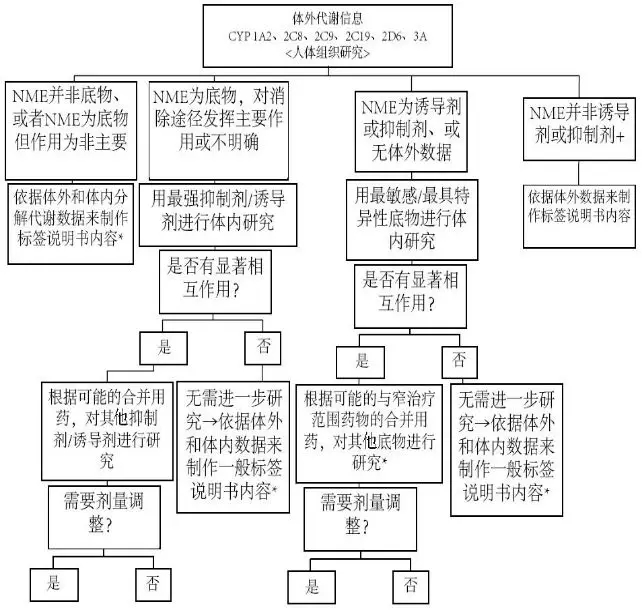
NME:新分子实体
* 补充的人群药代动力学分析结果可以帮助进行整体评价。
+ 测定NME是否为特殊CYP酶的抑制剂 或诱导剂 的标准;鸡尾酒研究阴性结果将排除测定NME是否为特殊CYP酶抑制剂或诱导剂的进一步研究 。
图 2、3提供了根据体外评价结果而对体内P-gp相互作用研究的决策树作为参考。
图 2.测定研究药物是否为P-gp底物、是否需要P-gp抑制剂的
体内相互作用研究的决策树
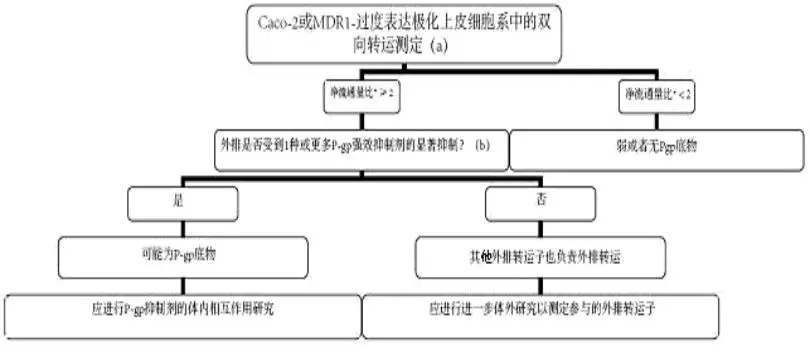
* Caco-2细胞的净流通量比计算公式为(通透性app, B-A/通透性app, A-B);对于MDR1-过度表达细胞系,净流通量比计算公式为(通透性app, B-A/通透性app, A-b)mdr1与(通透性app, B-A/通透性app, A-B)野生型的比值。
(a) 合格系统产生的探针底物的下一个净流通量比应与文献报告值相似。研究药物的净流通量比>2是需要进一步评价的阳性信号。注意:考虑到此值太宽泛,因此会产生过多阳性结果的担忧。还可以使用%值(研究药物相对探针底物(如地高辛)的净流通量)。
(b) 流通量比值显著减少(> 50%)或接近一致。
图3:测定研究药物是否为P-gp抑制剂、是否需要P-gp底物
(如地高辛)的体内相互作用研究的决策树
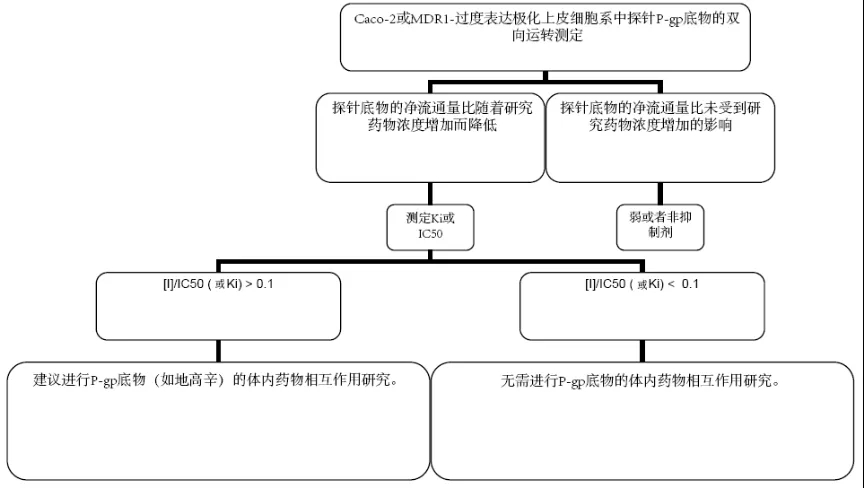
* Caco-2细胞的净流通量比的计算公式为(通透性app, B-A/通透性app, A-B);对于MDR1-过度表达细胞系,净流通量比的计算公式为(通透性app, B-A/通透性app, A-B)MDR1与(通透性app, B-A/通透性app, A-B)野生型的比值。[I]代表给予最高推荐临床剂量后、总药物(结合加未结合)的平均稳态Cmax值。
(二)特定的临床研究
通常在药物研发早期阶段进行适当设计的药代动力学临床研究,可提供有关代谢消除的途径、对总体消除的贡献和代谢相关的药物相互作用的重要信息。结合从体外研究获得的信息,这些体内临床研究可成为药物产品说明书陈述的主要基础,并有助于免除进一步的药物相互作用研究。
(三)群体药动学筛查
对大规模临床研究中,通过稀疏或密集采集血样所获得的数据进行群体药动学分析,这种分析对于探讨已知或新发现的相互作用的临床重要性,以及对于提出剂量调整的建议都可能非常有价值。如果通过临床研究数据分析检测到药物相互作用引起药物暴露量的重要变化,这些分析可以为相互作用提供参考信息,有时可以得到结论。群体药代动力学评价有可能发现非预期的药物相互作用。当存在早期的证据以及作用机制的数据时,群体药代分析还可以提供不存在某种药物相互作用的进一步证据。但是,如果专门设计用以评估药物相互作用的体外或体内研究的信息强烈提示存在某种相互作用,那么群体药代分析不太可能用以证明不存在这种相互作用。群体药动学研究的试验流程和样品采集均应经过严密设计,才能从中获得最多的信息。
四、体内药物相互作用研究设计
如果体外研究和其他信息提示需要进行体内代谢相关的药物相互作用试验,则应考虑下述一般性问题和方法。在下述讨论中,底物(S)一词用以表示确定其暴露量是否因使用另一种药物而改变的受试药物,而另一种药物称为相互作用药物(I)。根据研究目的,底物和相互作用药物可以是研究性新药(试验药)或已批准的药物。
(一)研究设计
体内代谢相关的药物相互作用研究通常是用于比较存在和不存在相互作用药物(I)的情况下的底物(S)浓度水平。研究可以使用随机交叉(如使用S后然后使用S+I,使用S+I后然后使用S)、同种序贯交叉(如使用S后总是使用S+I,或采用相反的顺序)、或平行设计(在一组受试者中使用S,另一组中使用S+I)等方式。对于底物和相互作用药物,可采用下述可能的给药方案组合:单剂量/单剂量、单剂量/多剂量、多剂量/单剂量、多剂量/多剂量。选择其中哪种试验设计将取决于底物和相互作用药物的许多因素,包括:(1)底物和/或相互作用药物的短期或长期使用;(2)安全性考虑,包括药物可能是窄治疗窗(NTR)或非窄治疗窗药物;(3)底物和相互作用药物的药动学和药效学特征;(4)诱导及抑制作用评估。具有抑制/诱导作用的药物和底物,用药时两种药物的暴露量应达到与其临床应用相关的血药浓度水平,包括可能使用的最高剂量。以下各点考虑可能有所裨益:
• 达到稳态很重要。但是当底物或相互作用药物和/或其代谢产物的半衰期较长、且不能使用可以迅速达到稳态的负荷剂量时,则需要使用特殊方法。这些方法包括选择同种序贯交叉或平行设计,而不选择随机交叉研究设计。
• 当相互作用药物为延迟效应,底物和/或相互作用药物需要在稳态下进行研究就非常重要,例如代谢酶的诱导剂和某些抑制剂介导的相互作用,证明相关药物以及感兴趣的代谢产物已接近达到稳态是非常关键的。该证据可通过先于相互作用试验前连续几天的采样研究而获得。尤其当代谢物的半衰期比原形药物长的情况下,达到稳态对代谢物及其原形药物均非常重要。如果原形药物和代谢物均为代谢抑制剂或诱导剂,这一点则更为重要。
• 研究通常可为开放试验(非盲设计),除非药效学终点对相互作用评估具有决定性作用(如不良事件评价容易出现偏倚时)。
• 对于一种迅速可逆的抑制剂,在试验当天,相互作用药物先于底物给药或与底物同时给药都可能增加研究的敏感性。对于机制性抑制剂(代谢后才能使酶灭活的药物,如红霉素),在底物给药之前给予抑制剂能使作用最大化。如果相互作用药物(如抑制剂或诱导剂)的吸收会受其他因素(如胃液pH)影响,那么控制其他变量并通过检测血浆内相互作用药物的浓度以证实其吸收情况是恰当的。
• 当关注两个药物对另一个药物的相互作用时,可以在单个研究或两个独立研究中评价潜在的相互作用,可选择随机三周期交叉、平行分组和同种序贯交叉设计。
• 在体内研究中,不受控制地服用食品添加剂、果汁或其他可影响不同代谢酶和转运体的食物可能导致研究结果不一致,所以在研究期间应避免食用这些食物是很重要的。
例如研究方案可以使用以下声明:“因以下原因可排除研究参加者:入选前两周内使用处方或非处方药物(包括中草药制剂)或酒精”、“研究开始前至少两周直至结束,志愿者将不允许食用或饮用任何含有酒精、葡萄柚或西柚汁、苹果或橙汁的食物或饮料,不得食用芥末科绿色蔬菜(如甘蓝、绿花椰菜、水田芥菜、绿色芥蓝菜、大头菜、包子甘蓝、芥末)和炭烧烤肉”。
(二)研究人群
临床药物相互作用研究通常采用健康志愿者。在健康人群的发现应当能够预测服用该药的患者人群的结果。基于安全性的考虑,有时不能采用健康受试者进行试验。然而,在某些特定情况下,患者人群更具有优势,这包括考察在健康受试者中不存在的药效学终点的机会。鉴定代谢相关遗传学多态性的表型或基因型,通常在评价具有多态性的酶系的影响时非常重要,特别是CYP2D6、CYP2C19和CYP2C9。药物相互作用(抑制或诱导)的程度可能不同,取决于特殊代谢酶的基因型。当受试者体内缺乏主要清除途径,比如未能显示出对药物的代谢,其他的途径就可能变得重要,此时应对其进行研究。
(三)底物和相互作用药物的选择
1.试验药为CYP酶的抑制剂或诱导剂
早期的研究方法主要集中于一组已批准的药物(地高辛、氢氯噻嗪),关注可能发生的合并用药、或相互作用导致的临床后果。随着对代谢相关的药物相互作用机制的进一步了解,可采用更通用的方法进行特定的药物相互作用研究,并获得更具普遍性的结论。在将试验药作为相互作用药物进行的研究中,体内研究最初的底物(已批准药物)选择取决于受相互作用影响的P450酶。在测试抑制作用时,一般应当选用某种与该酶系已知的特定抑制剂合并使用后,其药动学发生显著改变的底物,以评估试验药对底物的影响。底物举例:(1)咪达唑仑(CYP3A);(2)茶碱(CYP1A2);(3)瑞格列奈(CYP2C8);(4)华法林(CYP2C9)(评价S-华法林);(5)奥美拉唑(CYP2C19);(6)地昔帕明(CYP2D6)。如果最初研究结果显示研究药物可抑制或诱导代谢,那么根据合并用药的可能性,使用其他底物(代表一系列底物)进行进一步的研究可能是有意义的。如果最初研究结果显示对最敏感底物的抑制作用呈阴性,那么可以推测敏感性较小的底物也应不受影响。
CYP3A的抑制剂可根据合并用药后口服咪达唑仑或其他CYP3A底物在体内血浆中AUC的倍增变化进行分级。例如,如果试验药可使口服咪达唑仑或其他CYP3A底物的AUC增加5倍或更高(≥ 5倍)时,可标记为CYP3A强抑制剂。如果合用试验药(当以最高剂量及最短用药间隔给药时)可使口服咪达唑仑或其他敏感的CYP3A底物的AUC增加2-5倍(≥ 2- 而<5-倍)时,可将其标记为中等强度CYP3A抑制剂。同样,如果试验药(当以最高剂量及最短用药间隔给药时)可使口服咪达唑仑或其他敏感的CYP3A底物的AUC增加1.25- 2倍(≥ 1.25-而< 2-倍)时,可将其标记为CYP3A弱抑制剂。当试验药被确定为CYP3A抑制剂时,适当情况下,可在产品说明书中介绍其与敏感的CYP3A底物或治疗范围较窄的CYP3A底物的相互作用。
当体外评价不能排除试验药为CYP3A诱导剂的可能性时,可用最敏感底物(如口服咪达唑仑)进行体内评价。当给予多剂量试验药后同时口服咪达唑仑(可能已作为体内抑制作用评价的一部分),而结果呈阴性,那么可以推断出该试验药并非CYP3A的诱导剂(除了其并非CYP3A抑制剂的结论以外)的结论。经常采用口服避孕药进行体内诱导评价,然而,由于其并非最敏感底物,故阴性数据也不能排除试验药是CYP3A诱导剂的可能性。
在一项研究中同时给予志愿者CYP酶底物混合物(即“鸡尾酒(cocktail)方法”)是评价药物潜在抑制或诱导作用的另一种方法,但是需要对研究进行适当设计,同时应具备以下要素:(1)底物对各CYP酶具有特异性;(2)这些底物之间无相互作用;(3)对足量受试者进行了研究。鸡尾酒研究所得的阴性结果可以免除对个别CYP酶进行进一步评价的需要。然而,如果最初研究仅对尿液中原形药物与代谢物比值的变化进行了评估,那么所得阳性结果则表明需要进行进一步体内评价,以定量检测暴露量变化(如AUC、Cmax)。鸡尾酒研究的结果可用作其他评估药物对CYP酶抑制或诱导作用的体外和体内研究的补充数据。
2.试验药为CYP酶底物
考察试验药的代谢是否会被抑制或诱导(即作为底物)的试验中,相互作用药物应基于确定该药物代谢酶系的体外和体内研究结果来选择。相互作用药物可选用已知的、重要的代谢途径的抑制剂。例如,如果研究结果显示试验药经CYP3A代谢,而且该酶对试验药的总体消除的影响很重要(超过清除途径的25%)或未知,抑制剂和诱导剂可以分别选用酮康唑和利福平,因为它们在有关作用的鉴别中最敏感。如果研究结果为阴性,则表明该代谢途径不存在具有临床重要性的药物相互作用。如果采用最强有力的特异性抑制剂/诱导剂进行临床试验,结果显示阳性,而申请人希望确定试验药物与其他较弱的特异性抑制剂/诱导剂之间是否存在相互作用、或者对剂量调整提出建议,一般需要进行进一步的临床研究。如果一个药物经CYP3A代谢,且CYP3A抑制剂可使其血浆AUC增加5倍或更高,可认为该药物为CYP3A的敏感底物,产品说明书中应标明其为“敏感的CYP3A底物”,根据药物的暴露量-反应关系,该药物与强效或中等强度的CYP3A抑制剂合用时需注意。如果一个药物是经CYP3A代谢,且其暴露量-反应关系表明合用CYP3A抑制剂引起的暴露量水平增加可能会导致严重的安全隐忧(如尖端扭转型室性心动过速),那么可认为该药物为“治疗范围较窄的CYP3A底物”。
如果某个口服药物为CYP3A的底物,且因被小肠CYP3A广泛代谢而导致口服生物利用度很低,则西柚汁会对其系统暴露量产生显著影响。根据药物的暴露量-反应关系,该药物和西柚汁合用应格外注意。
如果药物为CYP3A或P-gp的底物,并且与贯叶连翘合用时将降低其系统暴露量和有效性,需将贯叶连翘以及其他已知的诱导剂(如利福平、利福布丁、利福喷丁、地塞米松、苯妥英、卡马西平或苯巴比妥)一同列于产品说明书中,因为这些药物同样也可能会降低血浆药物浓度。
如果药物经具有多态性的酶(例如CYP2D6、CYP2C9或CYP2C19)代谢,将慢代谢者与快代谢者的药动学参数进行比较,比较结果可代表该药和这些酶的强抑制剂的相互作用强度,可能无需再进行与强抑制剂的相互作用研究。当以上研究显示具有显著的相互作用时,可能有必要用弱抑制剂进行进一步研究。
在某些情况下,同时评价多种CYP抑制剂对药物代谢的影响非常有意义。例如,当同时符合以下条件时,可进行一种以上CYP抑制剂的相互作用研究:(1)该药物显示血药浓度相关的安全性问题;(2)该药物经多个CYP酶代谢清除;(3)剩余的或不被抑制的药物清除率的比例较低。在这种情况下,多种CYP-选择性抑制剂对药物血浆AUC倍增变化的影响可能远远大于单个抑制剂给药的影响。不确定程度取决于剩余清除率分数(分数越小,抑制剂的影响越大)以及抑制途径的相对清除率分数。然而,如果单个抑制剂的研究结果引起安全担忧(如禁忌证),则无需进行多种抑制剂研究。其他考虑可能包括药物与多种抑制剂合用的可能性。在研究多种抑制剂对药物暴露量的影响之前,非常重要的一点是首先应确定各个CYP抑制剂单个给药的作用并根据计算机模拟方法估计同时使用多种抑制剂的综合抑制效果。基于安全考虑,与多种抑制剂联合使用以评价系统暴露量倍增的研究,应当使用较低剂量的试验药。
对主要CYP酶以及CYP酶对摄取/外排药物的转运体同时产生抑制作用的影响与多种CYP抑制剂有同样的意义。例如,伊曲康唑和吉非贝齐合用时对瑞格列奈系统暴露量(AUC)的显著影响可能由于酶和转运蛋白的集合效应。
3.试验药为P-糖蛋白的抑制剂或诱导剂
在测试试验药是否为P-gp抑制剂或诱导剂的试验中,选择地高辛或其他已知的P-gp底物是恰当的。
4.试验药为P-糖蛋白底物
在测试试验药的转运是否会被抑制或诱导(作为P-gp的底物)的试验中,应对P-gp抑制剂(如利托纳韦、环孢菌素或维拉帕米)或诱导剂(如利福平)进行研究。当药物同时也是CYP3A底物时,需用兼为P-gp和CYP3A的强效抑制剂(如利托纳韦)进行抑制作用研究。
5.试验药为其他转运体底物
在测试试验药的体内处置被抑制或诱导的可能性时(如非P-gp的底物,或除了为P-gp的底物外,还是其他转运体的底物),可适当使用某种多种转运体(如P-gp、OATP)的抑制剂,如环孢菌素。新近有关转运体底物药物(非P-gp的底物,或除了为P-gp的底物外,还是其他转运体的底物)的相互作用研究还包括某些HMG Co-A还原酶抑制剂,如瑞舒伐他汀和普伐他汀。
(四)给药途径
代谢相关的药物相互作用研究中给药途径很重要。一般应选择计划用于临床的给药途径。当研究药物被开发成多种给药途径时,是否有必要针对所有给药途径进行代谢相关的药物相互作用研究,应当根据预期的相互作用机制以及相对应的原形药物和代谢物的浓度-时间曲线的类似性而决定。如果将来只销售口服剂型药物,那么一般情况下不需要进行与静脉注射制剂相互作用的研究;当然从口服药物和静脉注射药物研究所得的信息对于了解在药物相互作用的总体效应中,吸收和/或系统前清除率对相互作用效应的相对贡献率的大小可能是很有用的。有时,某些给药途径可能会减少信息的有用性。例如,如果肠内CYP3A活性可以明显改变底物的生物利用度,那么底物的静脉给药就不可能显示出底物药物的相互作用。将已批准药物用作底物或相互作用药物时,给药途径取决于当前的上市制剂。
(五)剂量选择
对于底物(试验药或已批准药物)和相互作用药物(试验药或已批准药物)所进行的试验应当以最大的可能性发现相互作用。建议采用试验计划中或已批准的相互作用药物(作为抑制剂或诱导剂)的最大剂量和最短给药间隔。例如,当使用酮康唑作为CYP3A抑制剂时,选择以400 mg/天的剂量、给药数天,就比使用较低的剂量更好。当使用利福平作为诱导剂时,选择以600 mg/天的剂量、给药数天,就比使用较低的剂量更好。出于安全性的原因,在某些情况下,建议在研究中使用低于临床所用量的剂量。此种情况下,申请人应在方案和研究报告中对因使用较低剂量导致试验检测药物相互作用的灵敏度受限进行讨论。
(六)研究终点
药动学参数变化可用于评估药物相互作用在临床上的重要性。充分了解一般人群或特殊人群中预期的与非预期的药物效应中,剂量/浓度及浓度/效应之间的关系将有助于解释这些研究的结果。在某些情况下,除了药动学测定/参数以外,还可使用药效学终点,例如INR测定(当研究华法林相互作用时)或QT间期测定。
1.药动学终点
在每项研究中,应获得的底物药动学的指标和参数如下:(1)暴露量指标如AUC、Cmax、达到最大血药浓度的时间(Tmax)和其他适宜的指标;(2)药动学参数如清除率、分布容积和半衰期。某些情况下,这些指标对于抑制剂或诱导剂也可能有用,特别是评估两个研究药物之间可能的相互作用的研究。其他的指标可能有助于药物在稳态时的研究(比如谷浓度),以证明用药方法在相互作用发生之前和期间足以达到接近稳态的水平。在某些情况下,对于剂量、血药浓度水平和效应之间相互关系的理解可能使得人们尤其关注某些药动学指标和/或参数。例如,如果临床结果与达峰浓度密切相关(如肾上腺素受体激动药的心动过速作用),此时选择Cmax或其他早期暴露量指标就是最恰当的。相反,如果临床结果与吸收程度的相关性更大,那么就应该首选AUC。采样频率应该足以能够准确测定原形药物和代谢产物相关指标和/或参数。对于底物(无论是试验药还是已批准药物),测定其重要活性代谢产物的药动学均很重要。
2.药效学终点
通常情况下,测定药代动力学就足以进行药物相互作用研究,但药效学的测定有时也能提供额外的有用信息。当受关注的底物的研究终点的药代动力学/药效学关系尚未建立时,或当药效学变化现象并非仅仅是因药动学相互作用所导致(如奎尼丁和三环抗抑郁药对QT间期的叠加作用)时,需要进行药效学测定。大多数情况下,当将已批准药物作为研究中的底物时,应该从其他数据中获知由于药物相互作用使底物暴露量(Cmax、AUC)变化而造成其对药效学的影响。如果需要进行药代动力学/药效学研究,一般需要比经典药代研究规模更大的研究(如QT间期作用研究)。
(七)样本量和统计学考虑
药物相互作用研究的目的是为了测定在相互作用药物存在的情况下,底物暴露量是否会出现增加或降低。如影响存在,则需要通过对药代动力学/药效学关系的理解,对Cmax和AUC变化的意义进行评估。
药物相互作用研究结果应以在含有相互作用药物(S+I)、不含相互作用药物(只有S)情况下观察到的药动学指标的几何平均数比值的90%置信区间进行报告。置信区间对观察到的S+I及单独S情况下系统暴露量指标比值的分布提供了一种估计,也是对这种相互作用强度的概率估计。相比之下,这些研究不适于进行显著性检验,这是由于较小的、持续出现的系统暴露量差异在统计学上可能具有显著意义(p < 0.05),但在临床上可能不相关。
当存在具有潜在重要的药物相互作用(如比较结果表明(S+I)时系统暴露量指标增大两倍(某些NTR药物会稍小)或更大)时,申请人应根据研究中试验药或已批准药物的剂量-效应和/或药代动力学/药效学关系,对药物相互作用的临床意义提供特定的建议。对于一个新药来说,比较困难的问题是评价试验药作为底物的药物相互作用影响。对于试验药具有的抑制或诱导作用,可将研究的主要结果加入至其他药物的产品说明书中抑制剂或诱导剂的列表中。药物相互作用的信息将成为研究结果报告、以及作为试验药或已批准药物的药品说明书中剂量、给药方案调整、注意事项、警告、或禁忌证有关建议内容的依据。
申请人可能希望在药品说明书上做一预计不会发生药物相互作用的特定声明。在这些情况下,申请人最好能对药物相互作用推荐特定的无效范围或临床等效性区间。无效范围表示在此区间内,系统暴露量的变化不具有临床意义。
定义无效范围的方法有两种:
方法1:无效范围是依据人群(组)平均剂量和/或浓度-效应关系、药代动力学/药效学模型和其他可获得的底物药物信息以确定因无临床意义的药物相互作用导致的差异程度。如果药物-药物相互作用研究的系统暴露量指标的90%置信区间完全落在无效区间内,申请人可以得出结论,认为不会发生有临床意义的药物相互作用。
方法2:在没有方法1所定义的无效范围情况下,对于试验中使用的试验药和已批准药物,申请人可以采用无效范围的默认值,即80%~125%。当系统暴露量比值的90%置信区间完全落在80%~125%范围内时,通常可以认为未出现有临床意义的差异。然而,这是一个非常保守的标准,需要对足够的样本进行研究,以符合该标准。
对于一个特定的药物相互作用研究,受试者数量的选择将取决于以下因素:可检测或剔除其药物相互作用具有临床意义效应的最小值、个体间和个体内在药动学指标的差异、以及尚未认识到的其他因素或来源的差异性。
五、对产品说明书的相应要求
有关药物的代谢途径、代谢产物和药代动力学相互作用的所有信息应放在产品说明书中临床药理学部分的药代动力学内容中。药物代谢和相互作用的临床后果信息应酌情放在用法用量、药物相互作用、注意事项、禁忌症、或警告部分中。当某一章节已包括了临床后果的信息时,在其他的章节内就没有必要重复,而只须适当注释。当根据代谢途径或相互作用研究数据需要对剂量调整、禁忌证、或警告(如应避免合并用药)提出建议时,这些建议应该包括在用法用量、注意事项、禁忌症、或警告部分中。
在某些情况下,可列举一些不含本药的临床研究信息,并说明本药物预期可获得相似结果。例如,如果药物被测定为CYP3A强效抑制剂,则无需对所有CYP3A底物进行试验后方可对与CYP3A敏感底物和具有较窄治疗范围的CYP3A底物的相互作用提出警告。根据底物试验的实际结果,就可以在说明书中对所有敏感及具有较窄治疗范围的底物使用相同的产品提出警告。
如果药物被确定为CYP3A敏感底物或具有较窄治疗范围的CYP3A底物,则无需使用所有强效或中效CYP3A抑制剂进行试验来警告与强效或中效CYP3A抑制剂的相互作用。如果其代谢途径主要是通过CYP3A,则可在未对其进行任何实际试验研究的情况下写入说明书。同样,如果药物被确定为CYP3A敏感底物或具有较窄治疗范围的CYP3A底物,则无需使用所有CYP3A诱导剂进行试验以警告与CYP3A诱导剂的相互作用。
六、参考文献
1. Tucker G, Houston JB, and Huang S-M, “Optimizing drug development: strategies to assess drug metabolism/transporter interaction potential — toward a consensus,” Clin Pharmacol Ther. 2001 Aug;70(2):103-14; Br J Clin Pharmacol. 2001 Jul;52(1):107-17; Eur J Pharm Sci. 2001 Jul;13(4):417-28; Pharm Res. 2001, Aug;18(8):1071-80.
2. Bjornsson TD, Callaghan JT, Einolf HJ, et al., “The conduct of in vitro and in vivo 1593 drug-drug interaction studies, A PhRMA perspective,” J Clin Pharmacol. 2003, 43: 443-469, 2003.
3. Yuan R, Madani S, Wei X, Reynolds K, and Huang S-M, “Evaluation of P450 probe substrates commonly used by the pharmaceutical industry to study in vitro drug interactions,” Drug Metab Dispos. 2002, Dec;30(12):1311-9.
4. Huang, S-M, Hall, SD, Watkins, P, Love, LA, Serabjit-Singh, C, Betz, JM, Hoffman, FA, Honig, P, Coates, PM, Bull, J, Chen, ST, Kearns, GL, Murray, MD, “Drug interactions with herbal products & grapefruit juice: a conference report,” Clin Pharmacol Ther. 2004, 75:1-12.
5. Huang S-M, Lesko, LJ, “Drug-drug, drug-dietary supplement, and drug-citrus fruit and other food interactions — what have we learned?” J Clin Pharmacol. 2004; 44:559-569.
6. Advisory Committee for Pharmaceutical Science — Clinical Pharmacology Section meeting minutes April 20-21, 2003 (CYP3A classification and P-gp), Nov 17-18, 2003 (CYP2B6 and CYP2C8), November 3, 2004 (Drug interaction concept paper) http://www.fda.gov/cder/audiences/acspage/pharmaceuticalmeetings1.htm.
7. Lin JH, Yamazaki M, “Role of P-glycoprotein in pharmacokinetics: clinical implications,” Clin Pharmacokinet. 2003;42(1):59-98.
8. Zhang Y, Bachmeier C, Miller DW, “In vitro and in vivo models for assessing drug efflux transporter activity,” Adv Drug Deliv Rev. 2003, Jan 21;55(1):31-51.
9. Tsuji A, “Transporter-mediated Drug Interactions,” Drug Metab Pharmacokinet. 2002;17(4):253-74.
10. Zhang, L, Strong, JM, Qiu W, Lesko LJ, and Huang, S-M. Scientific Perspectives on Drug Transporters and Their Role in Drug Interactions [PDF] [external link] Mol Pharm. 2006; 3(1), 62-69, Epub Jan 4 2006.
11. CDER/CBER guidance for industry on Population Pharmacokinetics. That guidance document and other CDER guidances referred to in this guidance are available on the Internet at http://www.fda.gov/cder/guidance/index.htm.
12. Schuirmann, DJ, “A comparison of the two one-sided tests procedure and the power approach for assessing the bioequivalence of average bioavailability,” J Pharmacokin and Biopharm. 1987, 15:657-80.
13. Labeling for Human Prescription Drug and Biological Products — Implementing the New Content and Format Requirements
14. Shitara Y, Sato H, Sugiyama Y, Evaluation of drug-drug interaction in the hepatobiliary and 1639 renal transport of drugs, Annu Rev Pharmacol Toxicol 2005;45:689-723.
15. Guidance for Industry, Drug Interaction Studies —Study Design, Data Analysis, and Implications for Dosing and Labeling. http://www.fda.gov/.../Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf